BIOL 470: Gene tree and Coalescent simulations (Part 1)
Purpose
Most phylogenetics today uses DNA sequences to infer relationships among lineages, both within and between species. The purpose of these simulations is to supplement your readings about using gene trees phylogenetics. The suggested order of activities is:
- Read Emlen and Zimmer Chapter 8 up through section 8.2 (pg. 271)
- Work through simulations here
- Read blog post and paper applying coalescent methods and phylogenies to COVID-19 (see this post for links to readings)
Simulating Gene trees
Unless we’re interested in organisms where it’s impossible to recover DNA (e.g. old fossils), most modern phylogenetics uses DNA sequences (genes) to infer evolutionary relationships within and between species. The textbook has lots of static images showing how gene trees are used to infer relationships. Here, I use dynamic simulations to make these processes come to life and see how the static images emerge. I assume you have read the beginning of Chapter 8 and do not cover all terms here.
Assumptions behind simulations
The real biological world we’re trying to understand is complex, but it’s helpful to make some simplifying assumptions as we build our understanding of evolutionary theory. Note, these are not assumptions of all phylogenetic approaches, just assumptions I’m using here for illustration.
Assumption 1: constant (small) population size. We will run very small simulations of populations consisting of just a few individuals. The number of individuals is the same in every generation (constant population size).
Assumption 2: discrete generations. We will assume the populations evolves through discrete generations that don’t overlap (e.g. parents and offspring don’t live simultaneously).
Assumption 3: no selection. For simplicity, we’ll only track neutral alleles that have no affect on fitness. Hence, they only evolve by genetic drift. We’re also not considering migration from other populations right now. We will add mutation later.
Assumption 4: clonal reproduction of haploid organisms. We’ll also simplify things by assuming that haploid organisms reproduce clonally. This way we don’t have to track mating and genotypes; we can just focus on the dynamics of alleles.
We could relax all of these assumptions and get similar results. We’re making these assumptions to strip away unnecessary information and focus on the essential principles.
How to think about gene tree simulations
The textbook has several images that look something like this:
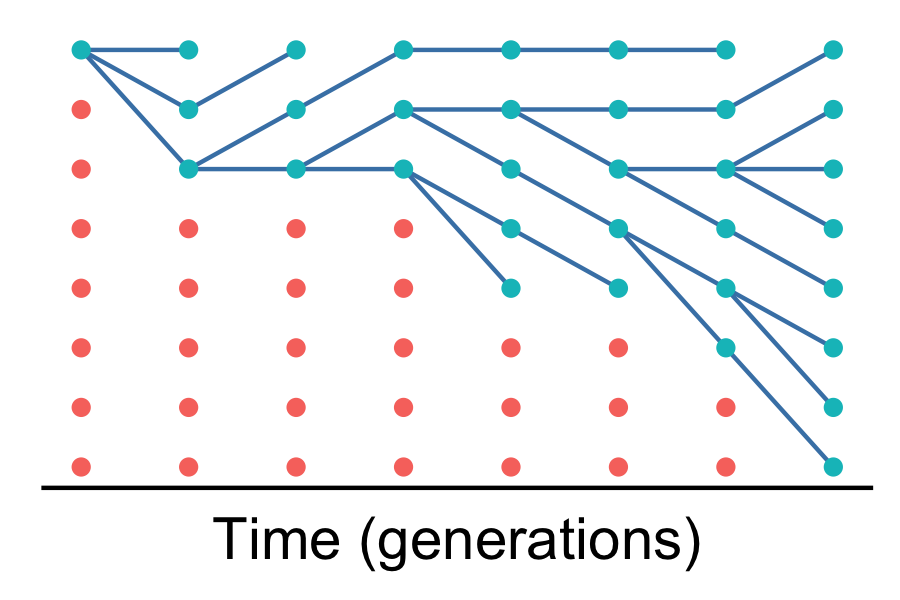
What’s going on here?
We’re focusing on one gene sequence at a single locus. Every gene tree in an organism’s genome will be different because of recombination.
We’re tracking the geneology of allele copies, not the frequency of certain alleles as we did in population genetics. This is an important, but challenging distinction.
Each column of 8 balls is the population in a single generation. Time proceeds from left to right. The right-hand side is today; the left-hand side is 7 generations in the past.
Within each generation (columns) there are 8 allele copies. Each allele copy is represented by a ball. We don’t yet know whether the sequences differ (we’ll get to that soon).
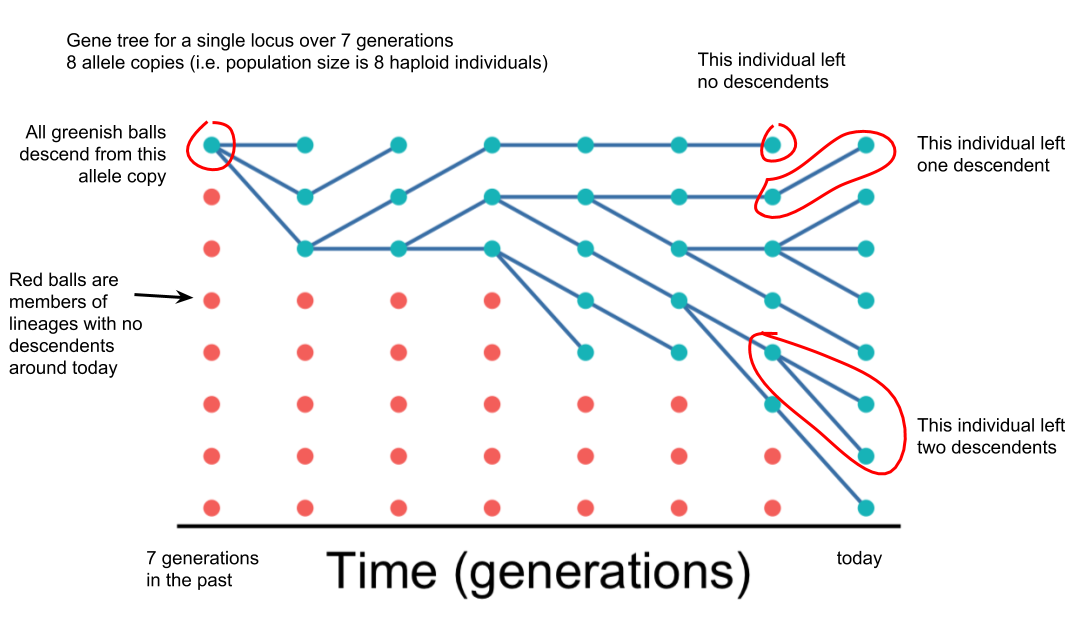
Lines between columns show descent (like a family tree). Some ancestors leave no descendents; other leave multiple descendents. However, this is all due to genetic drift because our simulation does not include selection.
All the living allele copies (greenish balls) descend from a common ancestor (the top-left corner).
Red balls indicate allele copies that left no descendents, so the lines of descent are not shown.
Also, remember this is a simulation. We “know” how every allele copy is related because we made up the data. Usually, we want to use DNA sequences to infer these relationships. Simulations (and the math behind them) tell us how to use DNA sequences to infer relationships.
Below I’ve annotated some key features of this gene tree
In this video, I’ll use coins and popsicle sticks to build a similar gene tree step-by-step. Apologies for the amateur production values.
Going from gene tree to a single-gene phylogeny
Real phylogenies often use several or even 1000s of sequences to infer relationships, but let’s start with a single gene phylogeny from our simulation above.
In the simulation, we can track the fate of allele copies that left no descendents. In the real world, we’re looking at relationships among living organisms and have no knowledge about these lineages that left no descendants. So the tree will just focus on the living organisms and cut out the rest. In the figure below, I’ve highlighted the nodes and lines of descent connecting them that will appear in our tree.
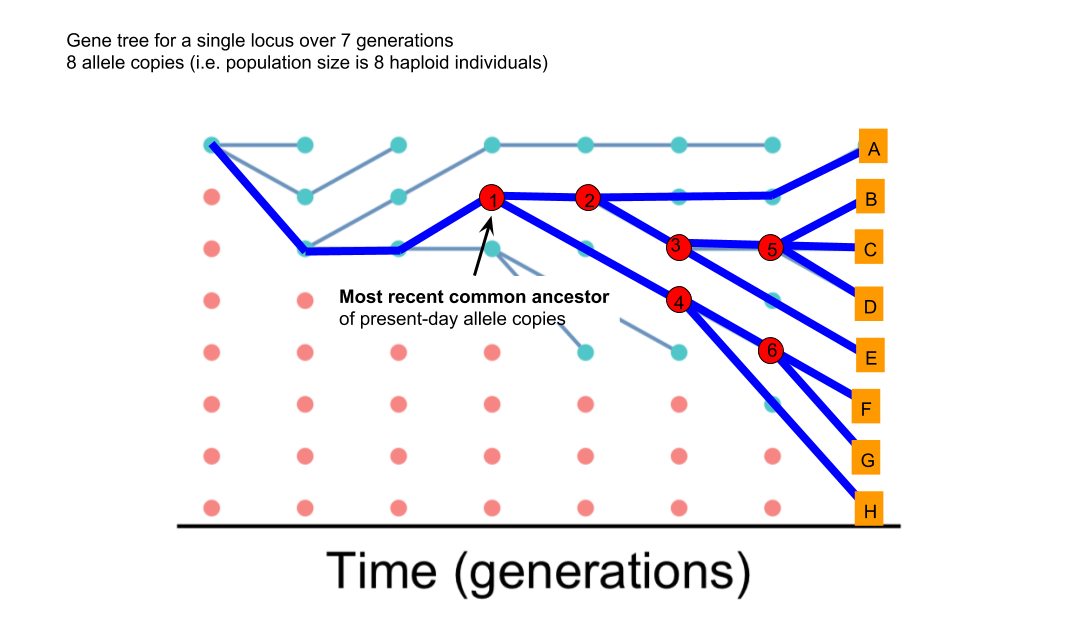
And here’s how the phylogenetic tree would look.
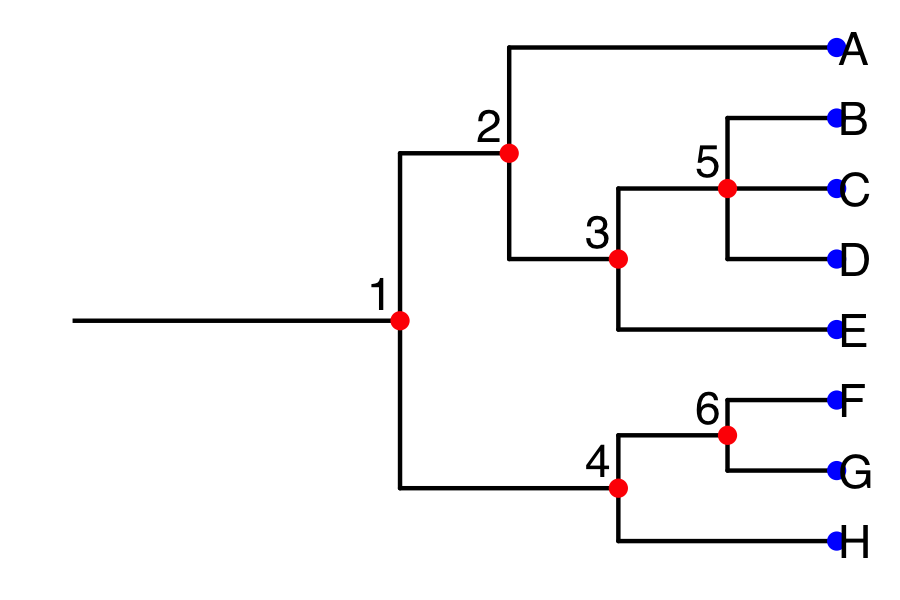
In simulations you do on your own, try going from a simulated geneology to a phylogeny depicting reletionships among the surviving allele copies at the tips.
Simulate on your own
Now that we’ve established the principles of gene tree simulations in a small populations, run simulations on your using this web app:
Try the activities described on the app page.